Angela Wood
Angela is well known as a leading expert in commercial and regulatory matters in the healthcare sector, with over 20 years' experience advising health, aged care, medical device and not-for-profit providers.
View profile
As part of the Therapeutic Goods Administration’s (TGA) regulatory reform agenda, the following new regimes are now in place:
This article outlines practical considerations for sponsors, manufacturers and suppliers of medical devices to support compliance with these new regimes. Select a topic from the list below to read more.
On 5 March 2025, the Procedure for Recalls, Product Alerts and Product Corrections (PRAC) replaced the Uniform Recall Procedure for Therapeutic Goods (URPTG). The Therapeutic Goods Administration (TGA) notes that the new procedure is intended to modernise and streamline the recall process for therapeutic goods, which includes medicines, medical devices and bloods/biologicals. While the PRAC looks wholly different to the URPTG - in substance, the actual process remains largely the same.
In summary, the PRAC involves:
The most significant difference between the URPTG and the PRAC is the simplification of terminology in the PRAC. In particular, the PRAC replaces the previous ‘recall’ and ‘non-recall’ actions with a single category of ‘market actions’ responding to the potential risk of harm involved.
Market actions are defined across three categories: class, type and level of market action, as described below.
The TGA determines the class of ‘market action’ by considering the severity of potential harm (critical, serious, minor or negligible) against the likelihood of that harm occurring (likely, sometimes, rarely or unlikely). Note that the “class” for market action purposes should not be confused with the “Class” classifications for medical devices.
The three classes of ‘market action’ are as follows:
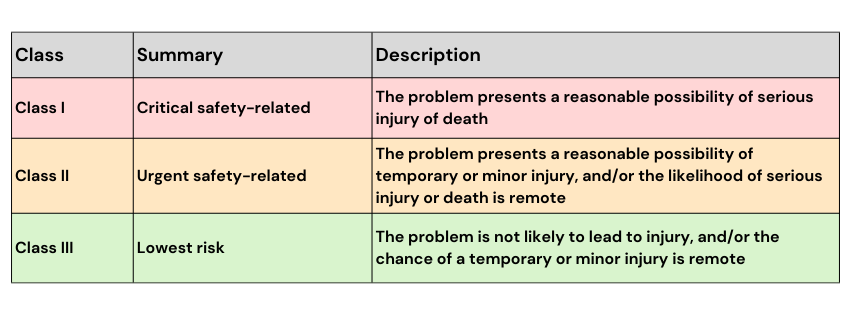
Source: Procedure for recalls, product alerts and product corrections (PRAC), TGA
The TGA has provided the following matrix to demonstrate how severity and likelihood are considered in determining the class of the market action:
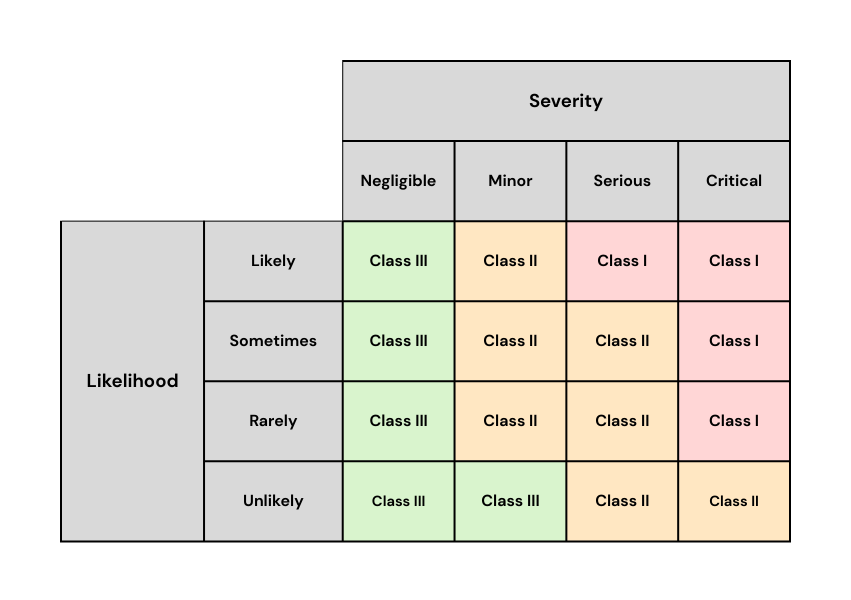
Source: Procedure for recalls, product alerts and product corrections (PRAC), TGA
The type of market action is the action that a sponsor or customer (such as a distributor) must take to address an actual or potential problem with the safety, quality, efficacy, performance, presentation or use of the medical device.
| There are four types of market action: | |
| 1. Recall: The permanent removal of the product from the market (and includes requesting customers to check and return defective products). | 2. Product Correction: The correction or fixing of specific or potential deficiencies in a product (e.g. repairs, modification, and updates to products). |
| 3. Product Alert: Alerting customers of certain or potential deficiencies, or general concerns relating to the use of a product. | 4. Quarantine: The temporary suspension of the use, supply or general distribution of the product, pending further investigation of the identified problem |
The level of market action determines who will be notified. The parties affected by market action have been retained from the URPTG: hospital, wholesale, retail and consumer levels. However the PRAC outlines considerations to assist in determining the appropriate communications about and channel of market action, which includes:
It is essential that sponsors, manufacturers and wholesalers/distributors have processes and systems in place to support compliance with the PRAC in the event of a requirement for market action, and that such parties have appropriate agreements in place to clearly delineate responsibility in the event of market action.
To assist, we have prepared the following compliance checklist:
| Responsible Party | Action | |
 | Sponsor | Review and update your Quality Management System (QMS) to ensure that it reflects the PRAC including:
in the event of market action, records of customer responses including procedures to follow up at least 3 times with customers who do not respond to the customer letter (not including the original dispatch of the letter). |
| Sponsor | Undertake appropriate internal training on the PRAC, including considering undertaking a mock market action. |
| Sponsor | Identify if any of your products are a ‘consumer good’ for the purposes of the Australian Consumer Law. If so, the ACCC must be notified of the market action within 2 days of receiving the agreement letter from the TGA. |
| Sponsors and manufacturers, wholesalers, distributors | Ensure that written agreements are in place between the sponsor and manufacturers and sponsor and wholesalers/distributors and the which appropriately set out the responsibilities of each party in responding to potential market actions, including:
|
| Sponsors, manufacturers and wholesalers/ distributers | Review processes and procedures to ensure that they support the ongoing and up-to-date collection and recording of relevant information including:
|
We previously reported that, in March 2023 the Therapeutic Goods Act 1989 (Cth) was amended to require healthcare facilities (being public hospitals, private hospitals and day hospitals) to report adverse events and near misses involving a reportable medical device. This reporting requirement was intended to commence on 21 March 2025.
Recent amendments to the Therapeutic Goods (Medical Devices) Regulations 2002 (Cth) have clarified the scope and commencement date of the mandatory reporting requirements, which will occur in three stages:
From March 2025, healthcare facilities may voluntarily report medical device-related adverse events as part of a 12-month transition period intended to allow time for facilities to build their reporting capability.
From 21 March 2026, healthcare facilities must report adverse events related to ‘reportable medical devices’, which are Class III or Class 4 IVD medical devices.
From 1 April 2028, the definition of ‘reportable medical device’ will be broadened to include all of the following:
The chief executive officer of a healthcare facility must give a report to the Secretary of the Department of Health, Disability and Ageing where any of the following occur:
| Therapeutic Goods Act 1989 (Cth) reference | Circumstances where a report is required | Time period to make the report |
| Section 41JM(2) | A reportable medical device is used in the facility and the use of the device results in the death or a serious deterioration in the health of a person while the device is used in the facility. | 10 days beginning on the day of the death of the person, or the day the serious deterioration in the health of the person is first identified. |
| Section 41JM(3) | A reportable medical device is not used in the facility because of the intervention of a person in that facility, and the use of the device would result in, or would likely result in, the death or a serious deterioration in the health of a person. | 45 days beginning on the day of the intervention. |
| Section 41JM(4). | A health practitioner provides treatment to a person in the facility for a serious deterioration in the health of the person, and the use of a reportable medical device has resulted in the serious deterioration in the health of the person. | 45 days beginning on the day of the provision of the treatment as mentioned. |
At a minimum, the reports must include:
Where a person is required to give a report to the Secretary but fails to comply with this requirement, the person will face a maximum civil penalty of 30 penalty units.
Healthcare facilities should use this 12 month transition period to ensure that their reporting processes are fit-for-purpose in providing the information required to the Secretary in the prescribed timeframe.
The TGA has indicated that it will release further guidance on reporting following the 12-month transition period.
Each medical device supplied in Australia has a UDI, which is a unique identifier for a medical device, comprising a:
On 24 March 2025, the TGA introduced the UDI regulatory framework to broadly align Australia’s identification system for medical devices with international approaches, with some local requirements. The UDI system aims to streamline the method of identifying medical devices to assist with post-market recall efficiency and traceability. UDI requirements will form part of the Essential Principles under the Therapeutic Goods (Medical Devices) Regulations 2002, meaning that manufacturers must comply with the UDI framework requirements to avoid criminal offences and civil penalties under the Therapeutic Goods Act 1989 (Cth).
The primary requirements under the UDI framework are:
To be comply, manufacturers must get their UDI-DI from a TGA recognised ‘Issuing Agency’.
Class I medical devices (non-sterile and non-measuring) are excluded from the mandatory UDI requirements, however manufacturers may choose to opt in.
Otherwise, all medical devices that must be included in the Australian Register of Therapeutic Goods (ARTG) must comply with UDI requirements, unless otherwise exempt.
Following the voluntary transition period, key dates for the commencement of mandatory UDI requirements are:
Impacted stakeholders including sponsors, manufacturers (existing or prospective) and healthcare facilities should be prepared for these changes and understand how these changes may impact them. Please get in touch if you wish to learn more.
The Prescription publication covers legal developments and trends in the healthcare and life sciences spaces in Australia.
Angela is well known as a leading expert in commercial and regulatory matters in the healthcare sector, with over 20 years' experience advising health, aged care, medical device and not-for-profit providers.
View profileKeep up to date with our legal insights and events
Sign up
Partner
Sydney